.png)
Gaucher disease (GD) is marked by extreme diversity in genotype, phenotype, age of onset, and disease severity, as well as an unpredictable, progressive disease course. Signs, symptoms, and clinical course may differ even among individuals with the same genotype and within the same family.1,2
Gaucher disease commonly mimics the signs and symptoms of many other hematological malignancies.1
Include Gaucher disease type 1 (GD1) in your differential diagnosis. These charts, broken out by specialty, may help.

Not all the signs and symptoms or potential differential diagnoses for Gaucher disease are included in this chart. Physicians should determine the appropriate differentials according to each patient’s condition.
*Acid sphingomyelinase deficiency (ASMD), historically known as Niemann-Pick disease types A, B, and A/B.
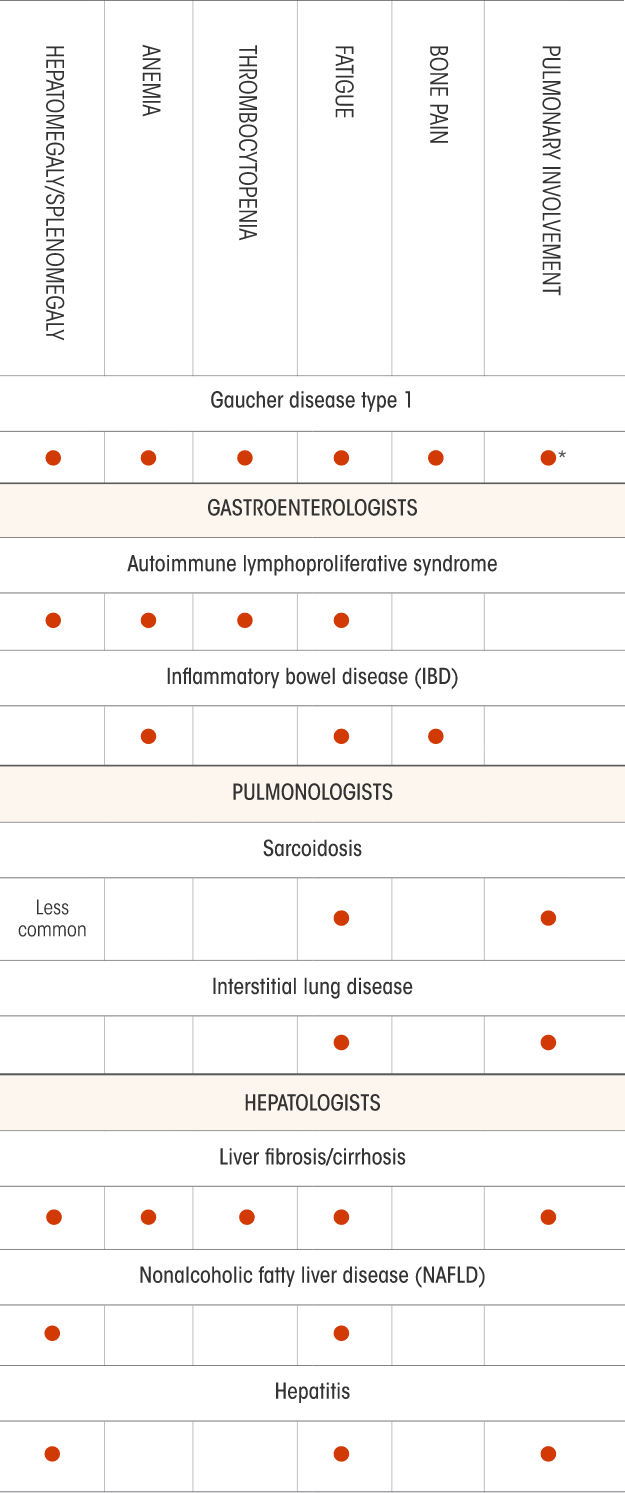
Not all the signs and symptoms or potential differential diagnoses for Gaucher disease are included in this chart. Physicians should determine the appropriate differentials according to each patient’s condition.
*Pulmonary symptoms are uncommon for patients with Gaucher disease type 1.
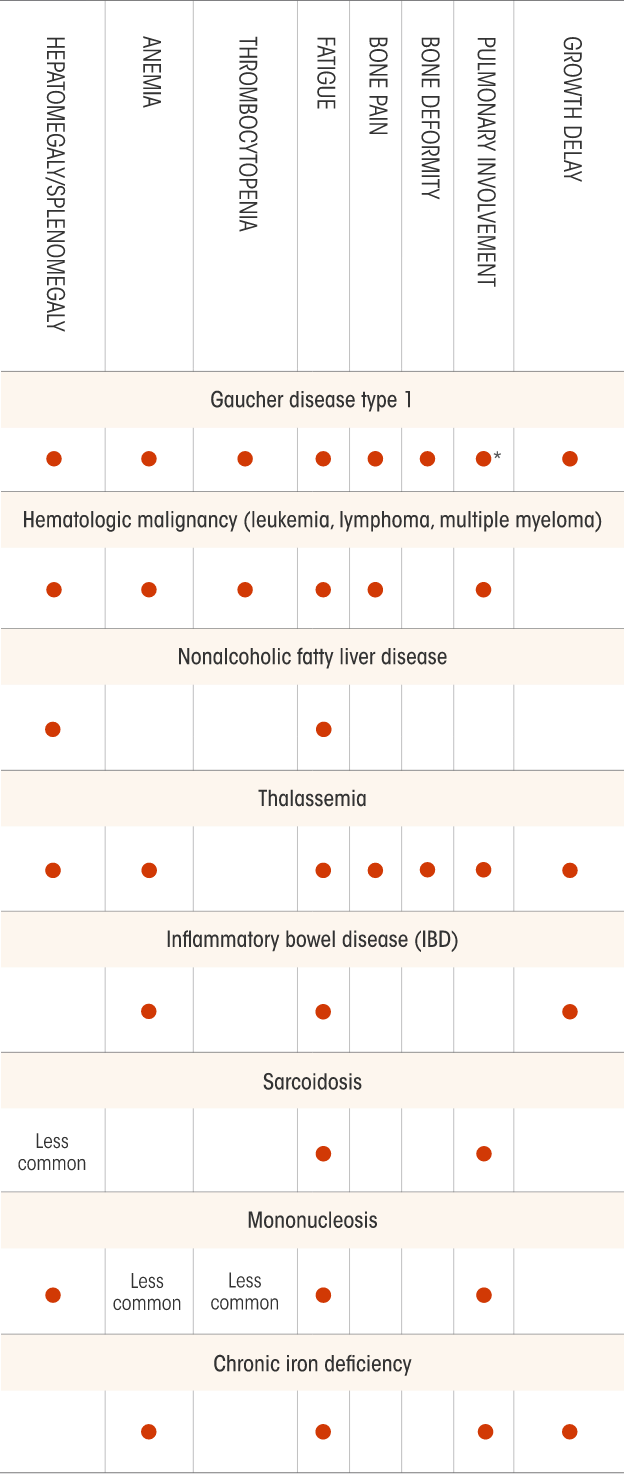
Not all the signs and symptoms or potential differential diagnoses for Gaucher disease are included in this chart. Physicians should determine the appropriate differentials according to each patient’s condition.
*Pulmonary symptoms are uncommon for patients with Gaucher disease type 1.

Early diagnosis and management of GD1 may help prevent irreversible complications or unnecessary procedures.1
Identify Gaucher disease with a blood-based enzyme assay3
Splenomegaly (87%) and thrombocytopenia (56%) are among the 2 most prominent and frequent presenting symptoms of GD.3,33
In non-Ashkenazi Jewish patients presenting with these symptoms, consider GD in your differential diagnosis once malignancy has been ruled out.3
It is recommended to test for GD as a first-line investigation in any patient of Ashkenazi Jewish heritage presenting with splenomegaly and thrombocytopenia.3
This diagnostic algorithm may help you know when to test for GD.

In patients of Ashkenazi Jewish heritage, the incidence of GD1 is higher (~1:850) than the incidence of hematologic malignancies (~1:2500).
Adapted from Mistry PK et al. Am J Hematol. 2011;86(1):110-115.
Beta-glucosidase enzyme assay is the standard, recommended method for establishing a confirmatory diagnosis of Gaucher disease, which is demonstrated by deficiency of beta-glucosidase activity.3
Molecular testing (DNA testing) can be used to establish a diagnosis of Gaucher disease, as well as carrier status.3
A bone marrow biopsy is typically not necessary or recommended to diagnose GD because although bone marrow biopsy specimens can rule out hematologic malignancies, they are not reliable for establishing the presence of Gaucher cells.3
DNA=deoxyribonucleic acid.
Gaucher disease type 1 management
GD1 may display inactive periods interrupted by episodes of acute crises or evidence of disease advancement. Patients may appear to have no symptoms, yet harbor mild disease manifestations such as cytopenia, splenomegaly, or osteopenia.2,3
GD1 requires an individualized disease management model that considers the progressive nature of the disease and severity of clinical manifestations. Generally, pre-emptive treatment for GD1 before irreversible complications occur can be more effective at achieving established therapeutic goals than a “watchful waiting” approach.3

The International Collaborative Gaucher Group (ICGG) Gaucher Registry (sponsored by Sanofi) is the world’s largest cooperative observational study on GD. The ICGG established the Gaucher Registry in 1991 as a longitudinal database to characterize the natural history of GD, evaluate long-term treatment outcomes, and optimize patient care.

The following recommendations for monitoring were published in 2004 by experts in the clinical management of GD who have served as advisors for the ICGG Gaucher Registry. Physicians should determine their patients’ assessments and the actual frequency of necessary evaluations according to each patient’s situation, including individualized therapeutic goals and routine follow-up.
Would you consider Gaucher disease in your differential diagnosis for these patients?
Treatment may improve symptoms and may help prevent certain long-term complications of Gaucher disease type 1.
References:
1. Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists–oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007;82(8):697-701.
2. Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41(4 Suppl 5):4-14.
3. Mistry PK, Cappellini MD, Lukina E, et al. A reappraisal of Gaucher disease–diagnosis and disease management algorithms. Am J Hematol. 2011;86(1):110-115.
4. Grabowski GA, Petsko GA, Kolodny EH. Gaucher disease. In: Valle D, Beaudet AL, Vogelstein B, eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw Hill; 2014: chap 146. Accessed July 17, 2023. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225546056
5. Hoffman R, Benz EJ Jr, Silberstein LE, Heslop HE, Weitz JI, Anastasi J. Hematology: Basic Principles and Practice. 6th ed. Elsevier Saunders; 2013.
6. O’Donnell MR. Acute leukemias. Cancer Network. Published June 1, 2016. Accessed July 17, 2023. https://www.cancernetwork.com/view/acute-leukemias
7. Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999;340(17):1330-1340.
8. Savage DG, Szydlo RM, Goldman JM. Clinical features at diagnosis in 430 patients with chronic myeloid leukemia seen at a referral centre over a 16-year period. Br J Haematol. 1997;96(1):111-116.
9. Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341(3):164-172.
10. Hairy cell leukemia facts (FS16). Leukemia & Lymphoma Society. Revised October 2013. Accessed July 17, 2023. https://www.lls.org/sites/default/files/file_assets/hairycellleukemia.pdf
11. Hairy cell leukemia treatment (PDQ®)—health professional edition. National Cancer Institute. Updated December 21, 2022. Accessed July 17, 2023. https://www.cancer.gov/types/leukemia/hp/hairy-cell-treatment-pdq
12. Adult non-Hodgkin lymphoma treatment (PDQ®)-patient version. National Cancer Institute. Updated December 29, 2022. Accessed July 17, 2023. http://cancer.gov/cancertopics/pdq/treatment/adult-non-hodgkins/Patient
13. Shankland KR, Armitage JO, Hancock BW. Non-Hodgkin lymphoma. Lancet. 2012;380(9844):848-857.
14. Myelofibrosis facts (FS14). Leukemia & Lymphoma Society. Revised November 2015. Accessed July 17, 2023. https://www.lls.org/sites/default/files/file_assets/FS14_Myelofibrosis%20Fact%20Sheet.pdf
15. Tefferi A. Primary myelofibrosis: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol. 2013;88(2):141-150.
16. Al-Farsi K. Multiple myeloma: an update. Oman Med J. 2013;28(1):3-11.
17. Shah D. Multiple myeloma clinical presentation. Medscape. Updated January 13, 2023. Accessed July 17, 2023. http://emedicine.medscape.com/article/204369-clinical
18. Kaplan P, Baris H, de Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013;172(4):447-458.
19. McGovern MM, Dionisi-Vici C, Giugliani R, et al. Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet Med. 2017;19(9):967-974.
20. Cox GF, Clarke LA, Giugliani R, et al. Burden of illness in acid sphingomyelinase deficiency: a retrospective chart review of 100 patients. JIMD Rep. 2018;41:119-129.
21. Medline Plus. Autoimmune lymphoproliferative syndrome. National Institutes of Health. Updated December 1, 2018. Accessed July 18, 2023. https://medlineplus.gov/genetics/condition/autoimmune-lymphoproliferative-syndrome/
22. Mayo Clinic. Inflammatory bowel disease (IBD). Accessed July 17, 2023. https://www.mayoclinic.org/diseases-conditions/inflammatory-bowel-disease/symptoms-causes/syc-20353315?p=1
23. Nunes H, Bouvry D, Soler P, Valeyre D. Sarcoidosis. Orphanet J Rare Dis. 2007;2:46.
24. Cleveland Clinic. Interstitial lung disease. Reviewed August 5, 2022. Accessed July 17, 2023. https://my.clevelandclinic.org/health/diseases/17809-interstitial-lung-disease
25. Mayo Clinic. Cirrhosis. Accessed July 17, 2023. https://www.mayoclinic.org/diseases-conditions/cirrhosis/symptoms-causes/syc-20351487?p=1
26. Mayo Clinic. Nonalcoholic fatty liver disease. Accessed July 17, 2023. https://www.mayoclinic.org/diseases-conditions/nonalcoholic-fatty-liver-disease/symptoms-causes/syc-20354567?p=1
27. Cleveland Clinic. Hepatitis: Viral hepatitis. Accessed July 17, 2023. https://my.clevelandclinic.org/health/diseases/4245-hepatitis-viral-hepatitis-a-b--c
28. Adams DH, Hubscher SG. Systemic viral infections and collateral damage in the liver. Am J Pathol. 2006;168(4):1057-1059.
29. Mayo Clinic. Thalassemia. Accessed July 17, 2023. https://www.mayoclinic.org/diseases-conditions/thalassemia/symptoms-causes/syc-20354995?p=1
30. Piga A. Impact of bone disease and pain in thalassemia. Hematology Am Soc Hematol Educ Program. 2017;2017(1):272-277.
31. Mayo Clinic. Mononucleosis. Accessed July 17, 2023. https://www.mayoclinic.org/diseases-conditions/mononucleosis/symptoms-causes/syc-20350328?p=1
32. Mayo Clinic. Iron deficiency anemia. Published January 4, 2022. Accessed July 17, 2023. https://www.mayoclinic.org/diseases-conditions/iron-deficiency-anemia/symptoms-causes/syc-20355034?p=1450
33. Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: Demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160(18):2835-2843.